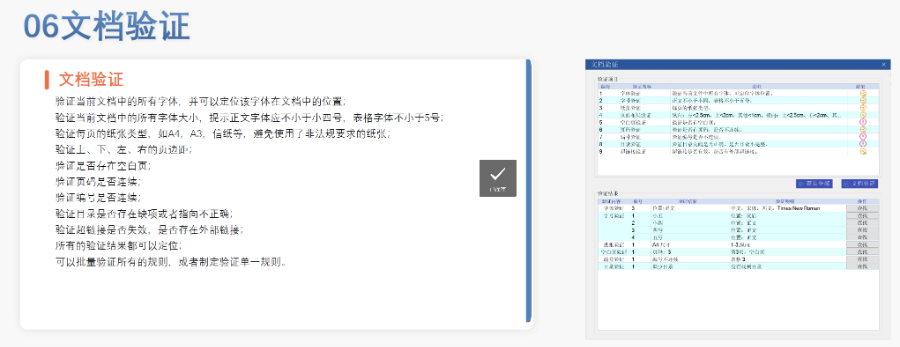
欧盟eCTD的递交途径与技术要求 不同审评程序对应不同递交渠道:集中程序(CP)通过EMA的eSubmission Gateway或Web Client提交,分散程序(DCP)和互认程序(MRP)则需使用欧盟通用提交门户(CESP)。文件结构需严格遵循模块化要求,例如CEP申请需包含模块1(行政文件)、模块2(质量概述)和模块3(技术文档),且XML主干文件须符合EDQM的特定命名规则。此外,所有PDF文件需无密码保护、可全文检索,并嵌入层级书签以支持快速审阅。 CEP申请的eCTD递交特殊性 CEP程序自2018年起强制采用eCTD格式,重点评估原料药是否符合欧洲药典标准。其模块1需包含EDQM申请表、简历及变更说明表,模块2需使用EDQM提供的质量概述模板,模块3则按CTD格式组织3.2.S章节内容。CEP与ASMF(活性物质主文件)的主要区别在于性:CEP无需关联上市许可,且审评由EDQM完成。欧盟eCTD验证标准相关技术支持。宁波新药eCTD报价

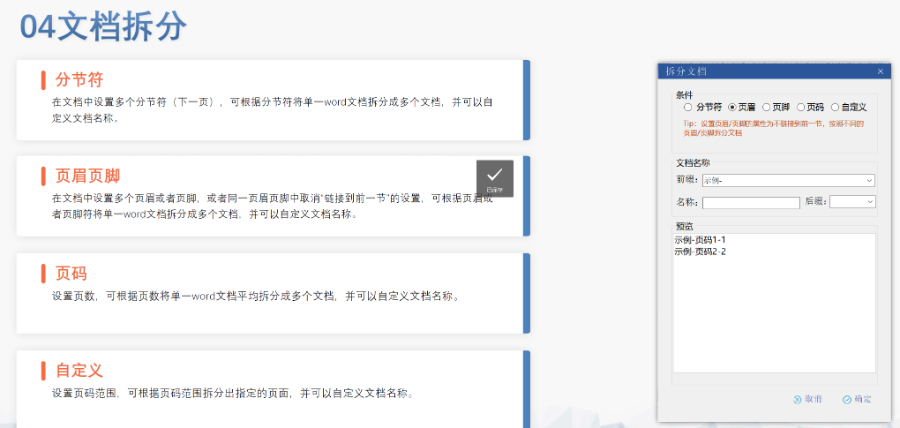
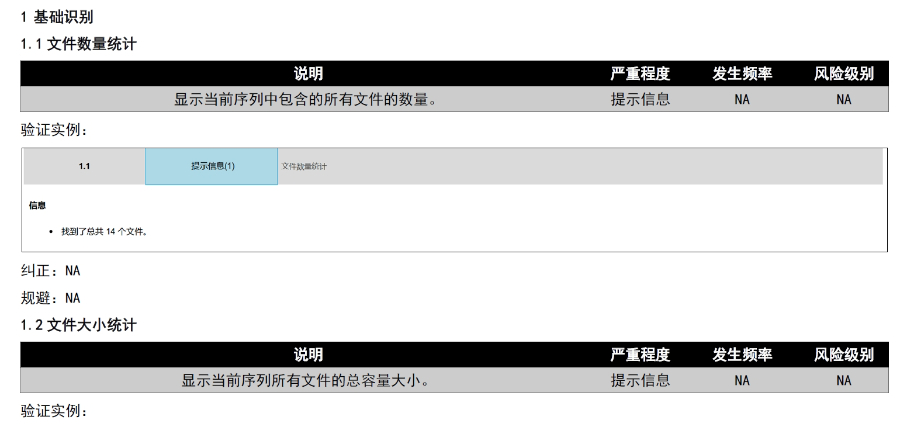
内容与格式检查Word预处理:需检查拼写、缩略语、单位格式(如),设置多级列表自动编号(如),统一字体(宋体/TimesNewRoman)和段落格式。重复内容处理:相同剂型不同规格可共用模块3,但需区分包装系统(如、)。外文资料:中文在前、原文在后,参考文献需中英文对照并建立跨网页链接。使用符合ICH标准的eCTD编辑器自动生成XML骨架和MD5校验值,拖拽PDF文件构建结构树。序列管理:序列号从0000开始递增,每次提交需更新序列,生命周期状态(New/Replace/Delete)需在XML中明确标注。验证与递交:确保无验证错误(如书签缺失、超链接断链),通过ESG等电子通道传输,光盘封面需包含申请号和序列号。全生命周期管理版本:通过软件实现网页签入/签出、审批流程,支持历史版本追溯。变更管理:增补(Append)和替换(Replace)需关联原始序列,删除(Delete)需彻底移除无效文件。 江苏中国eCTD医疗科技欧盟DMF注册申报相关技术支持。
此次eCTD实施范围的扩大对外企而言影响。实施范围的扩大为外企提供了更多选择,特别是在产品线中NDA和AND占比相当的情况下。外企的系统和流程相对成熟,因此它们对eCTD扩大范围持积极态度,更愿意尝试和改变。尽管过程中可能遇到技术或法规上的问题,但企业认为通过不断反馈和与CDE沟通,能够帮助提高整体申报效率和质量。此外,外企还面临向更集成化法规信息管理系统的挑战,特别是当需要迁移到系统时。如果尽早将产品迁移到eCTD,无论是系统迁移还是后续的生命周期管理都会更加顺畅。 随着eCTD实施范围的扩大,供应商将拥有更多的业务机会。然而,中国药品注册体系相对年轻化,在推进eCTD实施过程中可能面临各种问题。短期内,中小企业可能面临资金压力,需要考虑是否投入资金购买的eCTD系统。中长期来看,企业更关注的是如何建立一套完善的文档管理体系,而不是完成递交。这需要企业在前期投入更多时间和精力进行流程优化和人员培训。
区域化差异与多国协作挑战 欧盟eCTD需兼容成员国特定要求,例如模块一的行政信息需符合各国语言和法规差异。互认程序(MRP)中,参考成员国(RMS)的评估报告需被其他成员国认可,若出现分歧需由CMDh协调或提交EMA仲裁。这种多层级审评机制要求申请人在文件准备阶段即考虑区域兼容性,避免后续流程延误。 eCTD4.0的探索与未来方向 ICH于2015年发布的eCTD4.0版本旨在简化目录结构、支持多产品类型(如医疗器械)申报,并增强生命周期管理功能。欧盟计划通过2024年试点逐步过渡至4.0,其扁平化文件组织方式有望减少重复提交并提升审评效率。然而,实施需解决现有系统兼容性及行业适应性问题。澳大利亚ANDA注册申报相关技术支持。
美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效的电子申报服务。自2006年启用以来,ESG已成为FDA接收电子监管材料的入口,每日处理上千份提交文件,涵盖上市前审批、上市后监管、临床试验数据、不良反应报告等多种类型。该系统通过数字证书加密和公钥基础设施(PKI)技术,确保文件传输的真实性、完整性和不可否认性,符合FDA对电子提交的严格合规要求。在技术层面,ESG具备强大的文件处理能力。2018年系统升级后,取消了单个文件8GB的限制,可支持高达35GB的大型文件提交,进一步满足复杂申报需求。此外,文件格式需遵循eCTD(电子通用技术文档)规范,包括模块化结构、PDF标准化和XML元数据整合,以确保全球监管机构兼容性。2025年3月28日起,FDA将启用新一代平台ESG NextGen,逐步替代现有系统,过渡期需关注兼容性和稳定性问题。美国API的DMF申报相关技术支持。合肥中国eCTD性价比
美国ANDA注册申报相关技术支持。宁波新药eCTD报价
危机应对与应急递交机制 在公共卫生紧急事件(如COVID-19)中,EMA允许简化eCTD序列,优先审评关键模块并暂缓非数据。申请人可通过快速通道(Fast Track)提交疫苗或药物的eCTD资料,审评周期可压缩至6个月。此类申请需附风险评估报告,并承诺后续补交完整数据。 数据安全与长期存档 欧盟要求eCTD资料存档期限至少为药品上市后30年,EMA采用分布式存储和区块链技术确保数据不可篡改。申请人需定期备份本地副本,并使用符合GDPR要求的加密传输协议(如AS2)递交。历史数据的迁移和格式转换(如NeeS转eCTD)需遵循特定技术规范。 环保效益与可持续发展 eCTD取代纸质递交后,欧盟每年减少约500吨纸张消耗,审评流程的数字化降低碳足迹约30%。虚拟审评会议和电子签名进一步减少了差旅需求,契合欧盟2050碳中和目标。未来,eCTD4.0将通过数据压缩技术进一步降低服务器能耗。宁波新药eCTD报价