-
 陕西加拿大eCTD04.16
陕西加拿大eCTD04.16欧盟eCTD的递交途径与技术要求 不同审评程序对应不同递交渠道:集中程序(CP)通过EMA的eSubmission Gateway或Web Client提交,分散程序(DCP)和互认程序(MRP)则需...

-
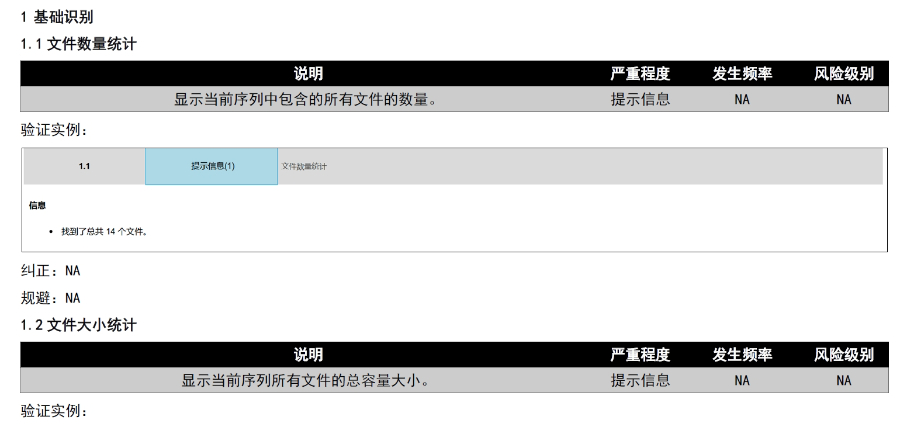 无锡CDE eCTD性价比高04.16
无锡CDE eCTD性价比高04.16仿制药作为提高药物可及性与可负担性的一类药物,2012年以前,注册审评是不收取任何费用的,但当时仿制药申请积压严重,从申报到获批需要3~5年的时间。 美国国会于2012年颁布了仿制药使用者费用修正案(...
-
 CDE eCTD报价04.16
CDE eCTD报价04.16DMF维护与合规 年度更 即使无变更,每年需提交声明;重大工艺/设施变更需及时通知客户并更文件。 现场检查 原料药企业需通过FDA现场检查,验证是否符合ICH Q7 GMP标准,并与DMF内容一致...
-
 浦东新区中国eCTD哪个品牌好04.16
浦东新区中国eCTD哪个品牌好04.16区域化差异与多国协作挑战 欧盟eCTD需兼容成员国特定要求,例如模块一的行政信息需符合各国语言和法规差异。互认程序(MRP)中,参考成员国(RMS)的评估报告需被其他成员国认可,若出现分歧需由CMDh...
-
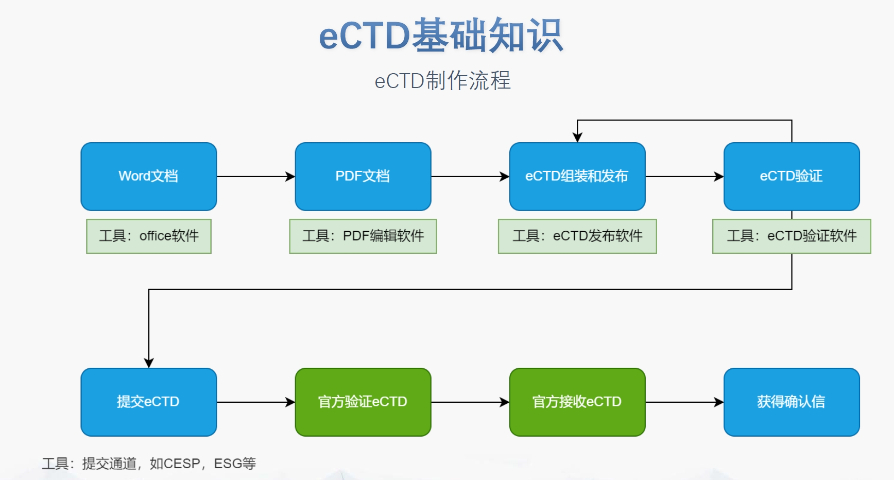 杨浦区CDE eCTD服务商04.16
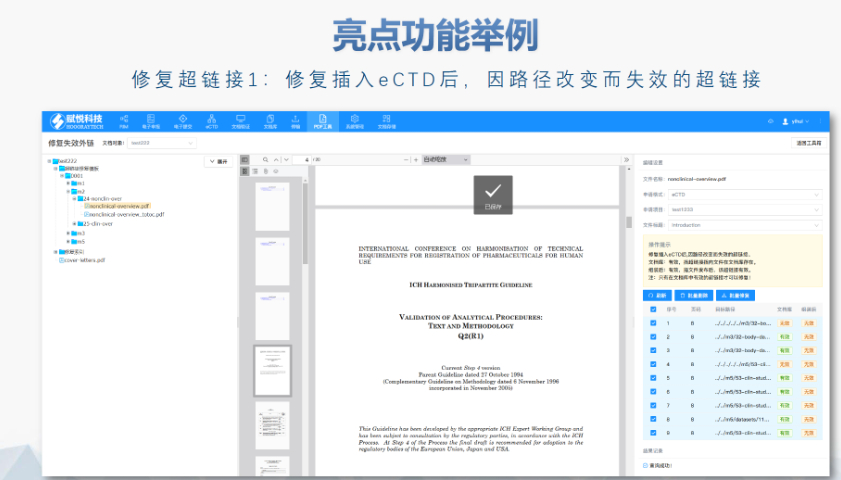
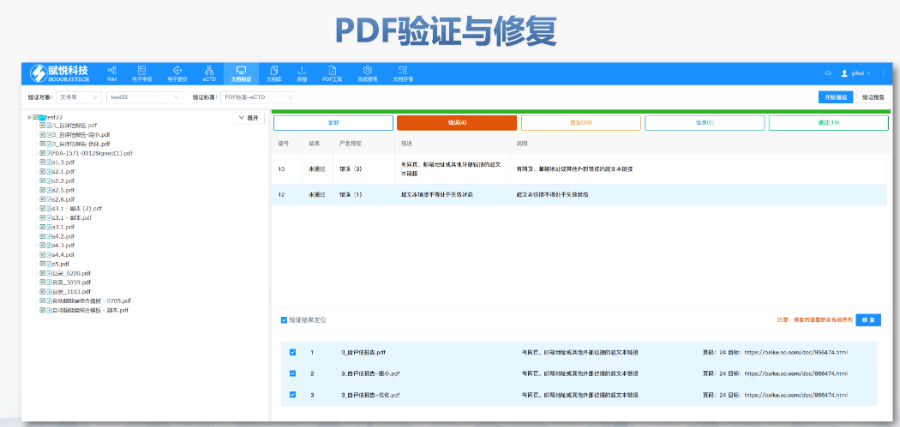
杨浦区CDE eCTD服务商04.16eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF...
-
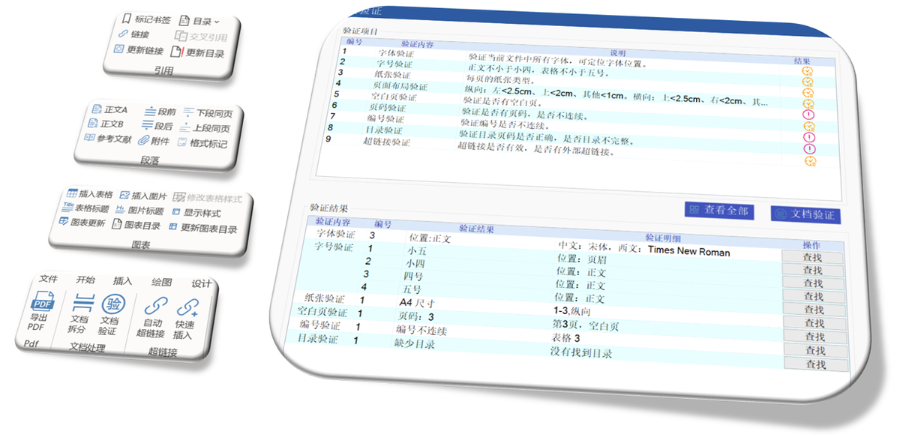 苏州NDAeCTD服务放心可靠04.15
苏州NDAeCTD服务放心可靠04.15美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效...
-
静安区原料药eCTD服务放心可靠04.15
eCTD的法规框架与技术规范:欧盟eCTD的法规层级包括指南(Guidelines)、指令(Directive)和法规(Regulation)。其中,法规(如CTR)具有直接法律效力,而指南(如ICH...
-
 芜湖中国eCTD哪家好04.15
芜湖中国eCTD哪家好04.15eCTD提交流程与ESG系统:FDA要求通过电子提交网关(ESG)传输eCTD文件,单个文件大小限制为10GB,超限需拆分或通过物理介质(如光盘)递交。提交前需预分配申请号(如NDA编号),并通过ES...
-
 工业园区中国eCTD系统04.15
工业园区中国eCTD系统04.15美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...
-
 合肥生物制品eCTD销售电话04.15
合肥生物制品eCTD销售电话04.15eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申...
-
闵行区国产eCTD是什么04.15
美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效...
-
 杭州化学药品eCTD04.14
杭州化学药品eCTD04.14ANDA递交: 按照ICH M4的CTD格式整理资料,并以eCTD格式递交; 通过ESG通道递交资料; 收到CDER的letter,说明资料已经进入FDA数据库; 付GDUFA费,在资料递交后的10日...