-
 太仓赋悦科技eCTD服务电话04.13
太仓赋悦科技eCTD服务电话04.13欧盟eCTD的历史沿革与强制实施 欧盟自2003年逐步推进eCTD(电子通用技术文档)的标准化进程,初要求药注册申请(MAA)采用CTD格式。2010年,集中审评程序(CP)率先强制使用eCTD,随后...

-
 无锡CDE eCTD哪个品牌好04.13
无锡CDE eCTD哪个品牌好04.13技术壁垒与兴市场挑战 非洲和东南亚国家逐步采纳eCTD,但其IT基础设施薄弱导致实施进度滞后。欧盟通过“eCTD全球化倡议”提供技术援助,帮助兴市场建立验证体系和培训中心。跨国药企需针对不同区域定制递...
-
 苏州中国eCTD供应商04.13
苏州中国eCTD供应商04.13经济影响与成本效益 尽管初期投入较高(平均每企业需50万欧元),但eCTD可减少30%的审评延迟成本,长期效益。仿制药企业通过eCTD复用原研数据,节省80%的申报准备时间。欧盟预算拨款2亿欧元资助中...
-
 静安区电子申报eCTD供应商04.13
静安区电子申报eCTD供应商04.13电子签章与安全性 FDA要求所有PDF文件需经数字签名,并通过MD5校验确保传输完整性。签章需符合21 CFR Part 11的电子记录规范,部分情况下允许临时放宽(如期间的远程签署)。 多模块协同...
-
 杭州国内注册eCTD性价比高04.13
杭州国内注册eCTD性价比高04.13欧美eCTD实施经验丰富,中国可借鉴以加速进程。中国可能会经历从企业自愿eCTD提交到强制eCTD提交的过渡,且将紧随ICH步伐,尤其在CMC资料整理方面。全球正向eCTD 4.0过渡,中国也不例外,...
-
 合肥国产eCTD文件如何制作04.13
合肥国产eCTD文件如何制作04.13欧盟eCTD的历史沿革与强制实施 欧盟自2003年逐步推进eCTD(电子通用技术文档)的标准化进程,初要求药注册申请(MAA)采用CTD格式。2010年,集中审评程序(CP)率先强制使用eCTD,随后...
-
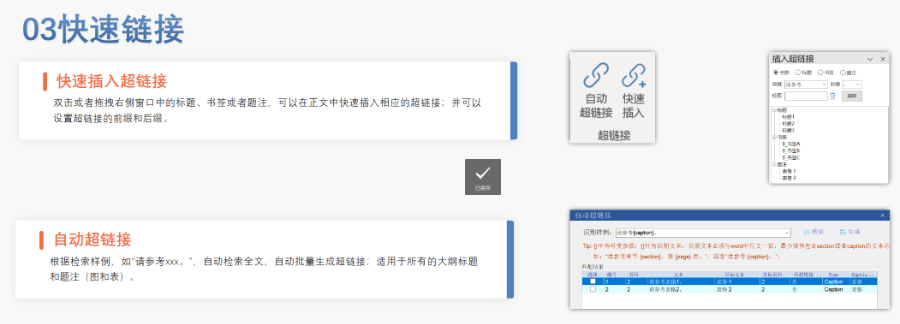 江苏新药eCTD是什么04.12
江苏新药eCTD是什么04.12仿制药作为提高药物可及性与可负担性的一类药物,2012年以前,注册审评是不收取任何费用的,但当时仿制药申请积压严重,从申报到获批需要3~5年的时间。 美国国会于2012年颁布了仿制药使用者费用修正案(...
-
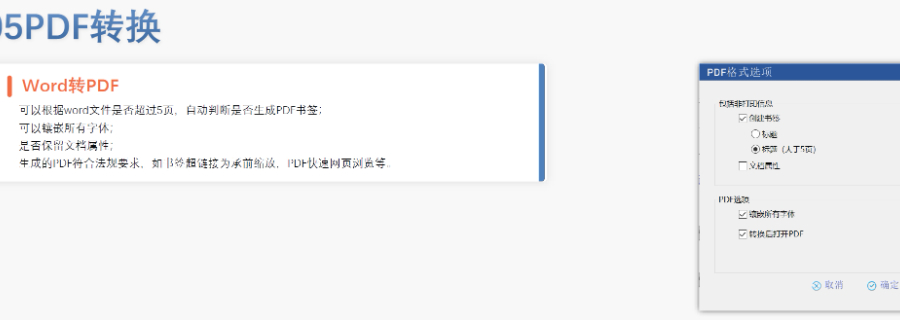 高新区化学药品eCTD发布系统04.12
高新区化学药品eCTD发布系统04.12eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF...
-
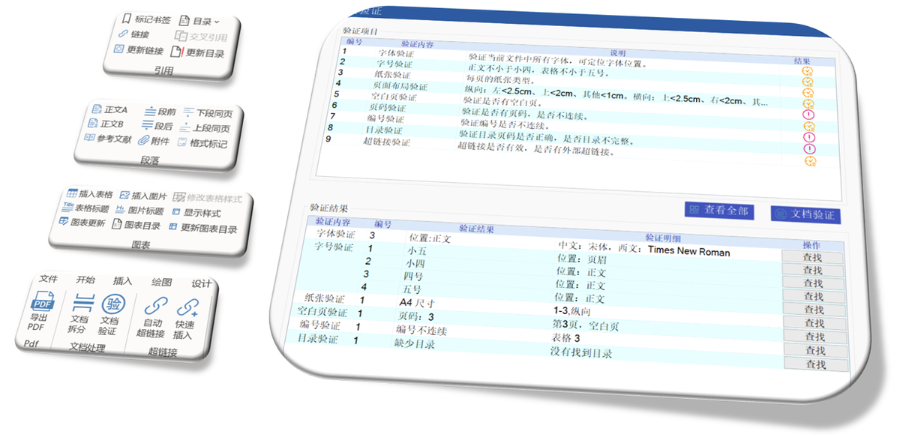 杨浦区NDAeCTD注册系统04.12
杨浦区NDAeCTD注册系统04.12多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA...
-
宁波赋悦科技eCTD服务介绍04.12
2015年发布《关于药品医疗器械审评审批制度的意见》,提出药监五大目标,将eCTD纳入国家药监数字化战略。2017年,中国加入ICH(国际人用药品注册技术协调会),成为全球第八个监管机构成员,加速与国...
-
无锡NDAeCTD常用解决方案04.12
经济影响与成本效益 尽管初期投入较高(平均每企业需50万欧元),但eCTD可减少30%的审评延迟成本,长期效益。仿制药企业通过eCTD复用原研数据,节省80%的申报准备时间。欧盟预算拨款2亿欧元资助中...
-
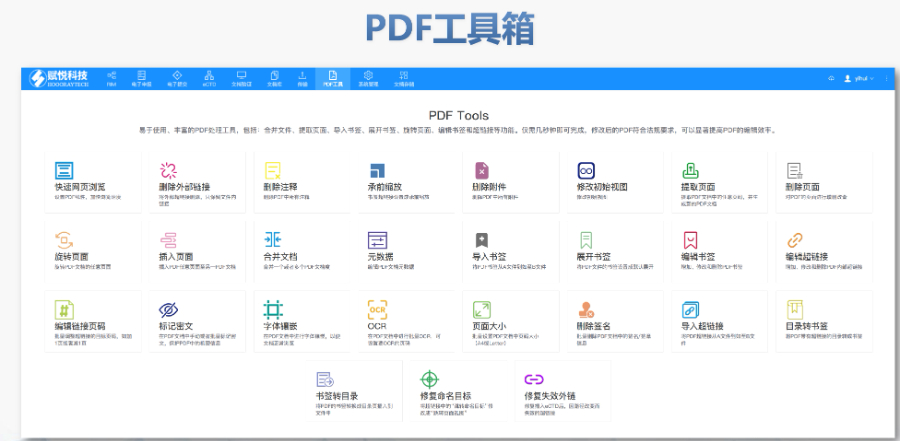 内蒙古eCTD哪个品牌好04.12
内蒙古eCTD哪个品牌好04.12GDUFA III框架与费用分类 2022年更的GDUFA III将费用分为ANDA申请费、DMF认证费、项目费及设施费四类,实施周期至2027年。2025财年ANDA费用涨至约22万美元,较2024...