技术壁垒与兴市场挑战 非洲和东南亚国家逐步采纳eCTD,但其IT基础设施薄弱导致实施进度滞后。欧盟通过“eCTD全球化倡议”提供技术援助,帮助兴市场建立验证体系和培训中心。跨国药企需针对不同区域定制递交策略,例如在模块1附加本地稳定性数据。 监管科学与创激励 eCTD支持真实世界证据(RWE)和适应性临床试验设计的整合,加速创药上市。EMA的PRIME计划为突破性疗法提供eCTD快速通道,允许分阶段提交模块数据。孤儿药和儿科药的eCTD序列可享受费用减免和优先审评。 供应链安全与审计追踪 eCTD的XML主干文件记录所有提交版本,支持供应链问题的追溯分析。原料药CEP持有者需及时更变更信息,确保下游制剂厂商获取数据。区块链技术试点用于追踪eCTD数据流,防止篡改和未授权访问。 文化差异与实施障碍 部分南欧国家偏好传统纸质流程,导致eCTD推广阻力较大。EMA通过多语种培训材料和区域协调员制度促进文化适应。行业需调整管理思维,将eCTD从“合规负担”转化为“竞争优势”。瑞士eCTD注册咨询相关技术支持。杨浦区ANDAeCTD服务介绍
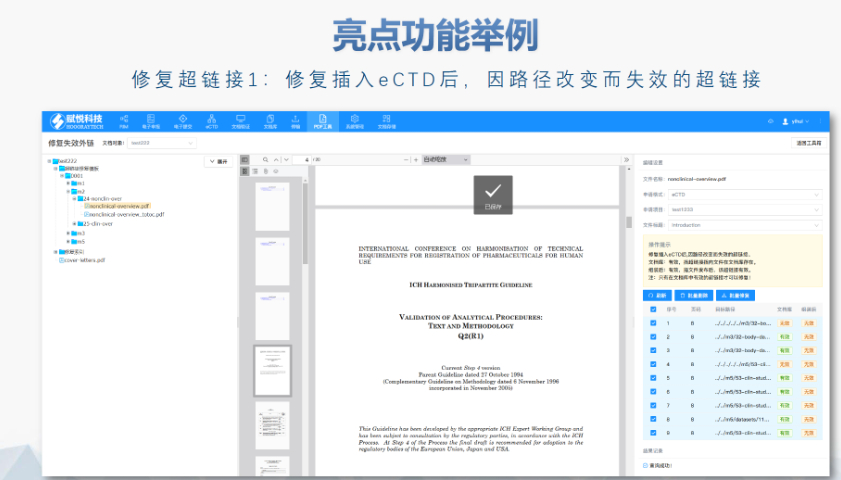
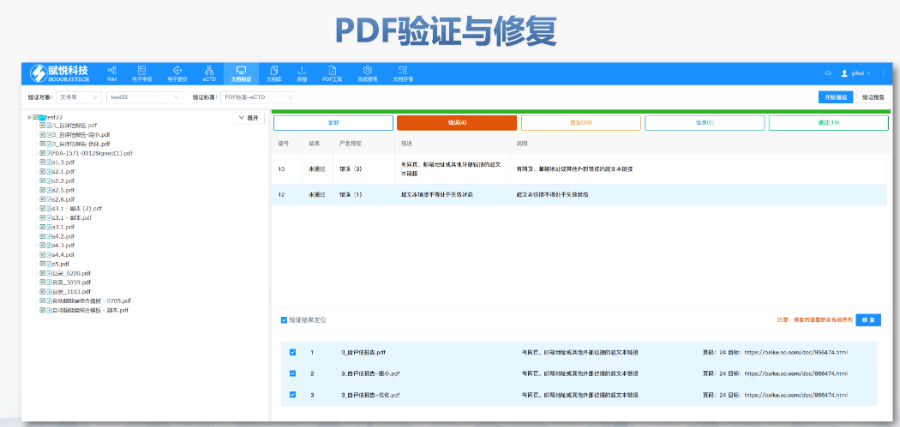
文件生命周期管理:eCTD支持文件替换(Replace)、删除(Delete)等操作,而非增文件。例如,更临床研究方案时需用Replace操作覆盖旧版本。基线提交(Baseline Submission)可用于补充历史纸质资料,但需在封面函中声明无内容变更。 临床数据与研究标签文件(STF):模块4和5中的研究数据需通过STF(Study Tagging Files)引用,确保数据与文档关联。FDA要求数据集(如SAS XPORT格式)能置于模块3-5,且单个文件超过4GB需拆分。2022年统计显示,58%的ANDA因研究数据技术拒绝标准(TRC)错误被拒。 电子签名与表格要求:FDA表格(如356h、1571)需使用数字签名,PDF文件禁止加密或设置编辑限制。电子签名需符合21 CFR Part 11规范,确保身份验证、不可否认性和数据完整性。 外包服务与系统解决方案:赋悦科技累计提交超2000份eCTD申请,外包可降低40%人工错误率。山东国产eCTD系统美国eCTD申报软件相关技术支持。
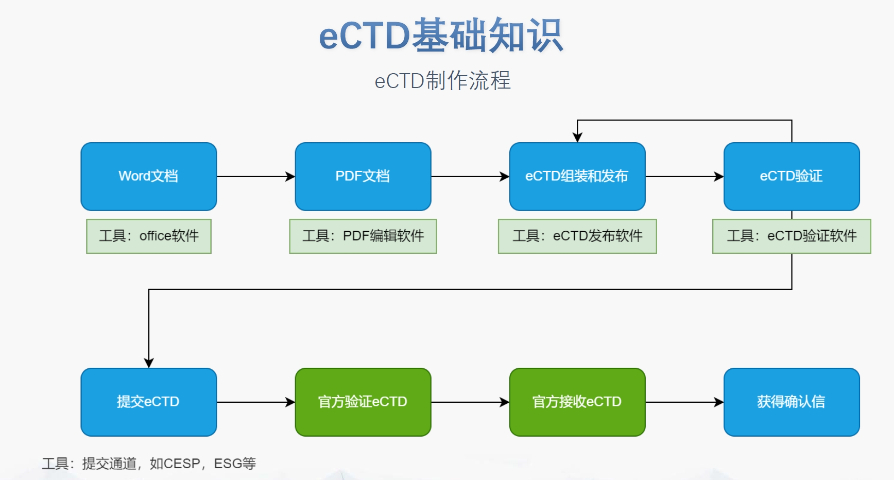
eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申报机制导致效率低下。2014年,欧盟通过《临床试验法规》(CTR, Regulation EU No. 536/2014),要求通过CTIS平台(临床试验信息系统)集中提交临床试验申请(CTA),并逐步推动eCTD作为电子化申报的工具。这一旨在解决传统CTD模式下审评周期长、多国协调成本高的问题,为eCTD的实施奠定了基础。
中国将进一步与国际接轨,推进eCTD等标准应用,提高药品注册效率和质量。AI技术可能在药品注册领域广泛应用,如辅助审评人员工作。未来药品注册资料将更注重结构化数据,有助于监管机构高效获取和利用数据。 eCTD等数字化工具将推动药品监管向智慧监管和全生命周期监管发展,提高监管效率和质量。区块链技术具有应用前景,可构建全球统一的药品申报数据平台。数据化时代,药品注册领域将更注重数据收集、分析和利用,为监管机构和企业提供决策支持。 总而言之,展望未来,随着eCTD在药品注册领域的广泛应用和不断发展,中国将逐步建立起与国际接轨的药品注册体系。这将有助于提高中国药品注册的效率和质量,推动中国药品走向世界舞台。同时,企业也需要密切关注技术发展动态和监管政策变化,及时调整自身战略和规划,以适应未来的市场竞争和监管要求。欧盟CESP提交通道相关技术支持。

GDUFA III框架与费用分类 2022年更的GDUFA III将费用分为ANDA申请费、DMF认证费、项目费及设施费四类,实施周期至2027年。2025财年ANDA费用涨至约22万美元,较2024年增幅达27.5%,反映审评成本上升。 ANDA申请费规则 费用需在提交时缴纳,若申请被拒可退还75%。重提交视为申请,需再次缴费。关联API的工厂数量影响总费用,例如某ANDA引用3个API且涉及6家工厂,需支付6倍DMF费用。 DMF费用机制 II类原料药DMF需在引用前缴费,一次性支付约5.3万美元(2025财年)。未缴费DMF不得用于支持ANDA,否则触发退审。 项目费分级管理 根据企业获批ANDA数量分为大、中、小型三级,2025年大型企业年费约34万美元。附属公司ANDA数量合并计算,缴费责任可由母公司或任一附属公司承担。加拿大DMF注册申报关技术支持。江西eCTD供应商
美国ANDA注册申报相关技术支持。杨浦区ANDAeCTD服务介绍
美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效的电子申报服务。自2006年启用以来,ESG已成为FDA接收电子监管材料的入口,每日处理上千份提交文件,涵盖上市前审批、上市后监管、临床试验数据、不良反应报告等多种类型。该系统通过数字证书加密和公钥基础设施(PKI)技术,确保文件传输的真实性、完整性和不可否认性,符合FDA对电子提交的严格合规要求。在技术层面,ESG具备强大的文件处理能力。2018年系统升级后,取消了单个文件8GB的限制,可支持高达35GB的大型文件提交,进一步满足复杂申报需求。此外,文件格式需遵循eCTD(电子通用技术文档)规范,包括模块化结构、PDF标准化和XML元数据整合,以确保全球监管机构兼容性。2025年3月28日起,FDA将启用新一代平台ESG NextGen,逐步替代现有系统,过渡期需关注兼容性和稳定性问题。杨浦区ANDAeCTD服务介绍