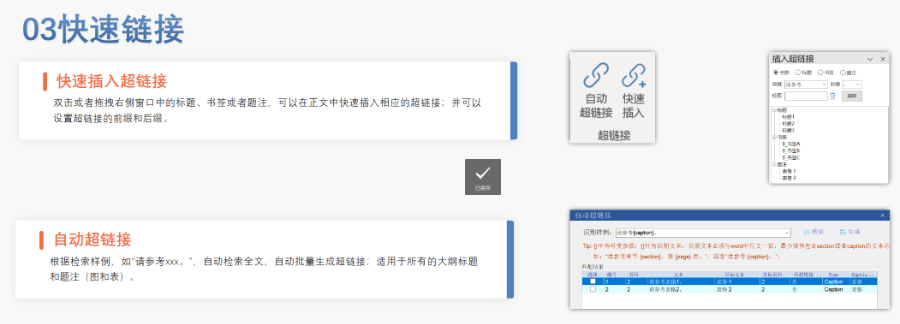
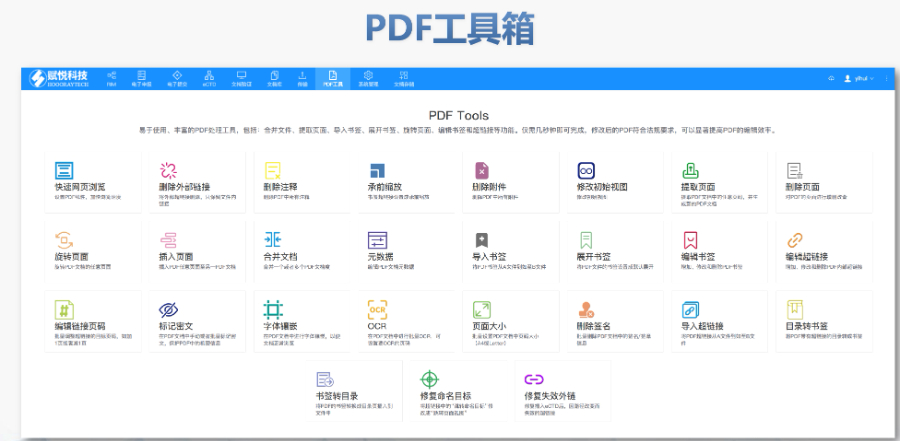
DMF维护与合规 年度更 即使无变更,每年需提交声明;重大工艺/设施变更需及时通知客户并更文件。 现场检查 原料药企业需通过FDA现场检查,验证是否符合ICH Q7 GMP标准,并与DMF内容一致。 转让与关闭 转让:需书面通知FDA并提供持有者信息。 关闭:未提交年度报告或持有人主动申请,需说明原因并通知所有授权方。 关键注意事项 数据质量:所有资料需准确、完整,减少审核延迟风险。 合规性:遵循FDA指南(如21 CFR Part 207)及USP标准(如培养基物料来源级别)。 沟通机制:建议通过专业机构(如瑞欧佰药)协助,定期提交周报并制定计划表以提高效率。 常见问题解答 生物制品分类:培养基、外泌体等均属Ⅱ类DMF。 质量标准:参考USP及同行标准,需提供分析方法验证及杂质对比研究。 周期估算:资料准备约5-50个工作日,总周期受缺陷回复影响。eCTD注册外包相关技术支持。安徽INDeCTD文件如何制作

eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申报机制导致效率低下。2014年,欧盟通过《临床试验法规》(CTR, Regulation EU No. 536/2014),要求通过CTIS平台(临床试验信息系统)集中提交临床试验申请(CTA),并逐步推动eCTD作为电子化申报的工具。这一旨在解决传统CTD模式下审评周期长、多国协调成本高的问题,为eCTD的实施奠定了基础。山西eCTD名称澳大利亚的eCTD申报相关技术支持。
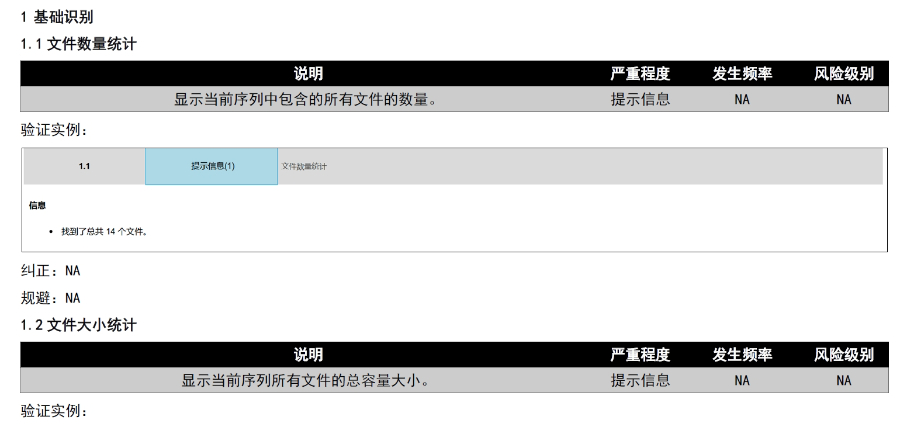
多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA的eSubmission Gateway提交,审评时限约240个工作日,eCTD需包含完整的模块1-5及多语言标签文件。 分散审评程序(DCP):需通过CESP(欧盟共同提交门户)递交,参考成员国(RMS)主导审评,eCTD需支持多国同步评估的模块化拆分。 互认程序(MRP):已授权成员国作为RMS,eCTD需包含基线序列(Baseline Sequence 0000)以整合历史审评数据,并通过CMDh协调分歧。
审评效率与时间线优化 eCTD的标准化缩短了审评周期:集中程序平均审评时间从18个月降至12个月,互认程序可在90天内完成成员国意见协调。自动化验证工具减少了格式错误导致的退审率,但复杂药学数据的科学审评仍需较长时间。申请人可通过预提交会议(Pre-submission meeting)提前沟通技术细节,规避潜在延误。 区域协作与全球互认 欧盟通过互认程序与澳大利亚、加拿大等国实现eCTD数据共享,CEP证书在40余个非欧盟国家有效。然而,模块一区域信息的差异性仍要求申请人定制化调整,例如亚洲国家可能要求附加稳定性研究数据。ICH的协调作用有助于减少重复提交,但完全全球化仍需解决法规和技术壁垒。 技术工具与行业生态 主流eCTD编辑软件(如Lorenz、Extedo)支持欧盟区域模板的自动化生成,并与验证工具集成实现一键校验。云平台解决方案逐渐普及,支持多国团队协同编辑和实时版本控制。然而,软件采购和维护成本较高,中小企业常选择外包给专业服务商完成递交。澳大利亚DMF注册申报相关技术支持。
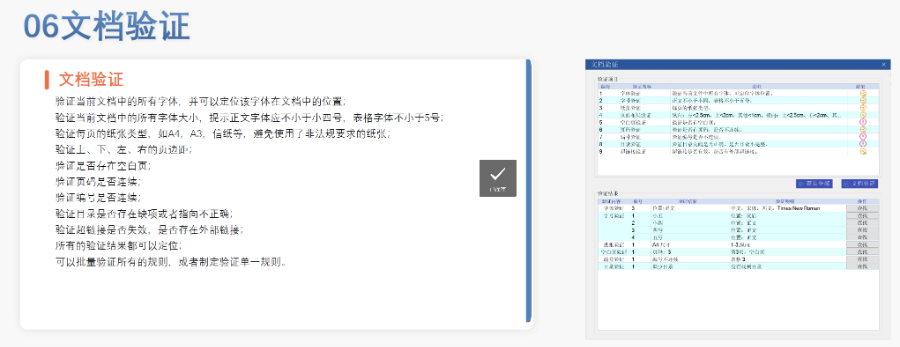
欧盟eCTD的历史沿革与强制实施 欧盟自2003年逐步推进eCTD(电子通用技术文档)的标准化进程,初要求药注册申请(MAA)采用CTD格式。2010年,集中审评程序(CP)率先强制使用eCTD,随后分散程序(DCP)和互认程序(MRP)分别于2015年、2017年跟进。至2019年,欧盟要求所有国家程序(NP)的注册申请均以eCTD格式提交,标志着其电子递交体系的成熟。2024年,EMA启动eCTD4.0试点项目,旨在提升技术兼容性与审评效率。 eCTD验证标准的迭代与关键更 欧盟的验证标准历经多次调整,例如2025年3月启用的eCTD3.1区域模板和验证规则v8.1,对文件结构、元数据和内容完整性提出更严格的要求。标准引入的“追踪表(Tracking Table)”强制校验规则(如15.11和15.12)曾导致CEP(欧洲药典适用性证书)递交,后通过允许占位文件临时解决。与早期版本相比,v8.1强化了对模块一区域信息的逻辑验证,并细化了对PDF书签、超链接的规范性检查。加拿大eCTD申报相关技术支持。浦东新区国际注册eCTD文件如何制作
加拿大ANDA注册申报相关技术支持。安徽INDeCTD文件如何制作
危机应对与应急递交机制 在公共卫生紧急事件(如COVID-19)中,EMA允许简化eCTD序列,优先审评关键模块并暂缓非数据。申请人可通过快速通道(Fast Track)提交疫苗或药物的eCTD资料,审评周期可压缩至6个月。此类申请需附风险评估报告,并承诺后续补交完整数据。 数据安全与长期存档 欧盟要求eCTD资料存档期限至少为药品上市后30年,EMA采用分布式存储和区块链技术确保数据不可篡改。申请人需定期备份本地副本,并使用符合GDPR要求的加密传输协议(如AS2)递交。历史数据的迁移和格式转换(如NeeS转eCTD)需遵循特定技术规范。 环保效益与可持续发展 eCTD取代纸质递交后,欧盟每年减少约500吨纸张消耗,审评流程的数字化降低碳足迹约30%。虚拟审评会议和电子签名进一步减少了差旅需求,契合欧盟2050碳中和目标。未来,eCTD4.0将通过数据压缩技术进一步降低服务器能耗。安徽INDeCTD文件如何制作